Background.
SARS-CoV-2’s Achilles’ heel has always been vitamin D in general and nitric oxide in particular. We first explained the relevance of vitamin D in July 2020, identifying the correlation between vitamin D deficiency and COVID-19 mortality rates and the key role of exposure to photosynthesis of vitamin D from exposure to UVB that occurs naturally from being in sunlight. In October 2020 we set out the two pathways – vitamin D→endothelial nitric oxide synthase→nitric oxide and cholecalciferol→calcifediol→calcitriol – through which the body produces one of the most, if not the most, effective countermeasures against SARS-CoV-2 infection and a proven compound to reduce COVID-19 disease progression and severity. It also has a function in preventing coagulation and clotting.
The relevance of nitric oxide is even greater as the ACE2→Angiotensin1-7→Mas receptor axis (so-called ‘good angiotensin’) increases NO production whereas its counterfunction, the ACE→angiotensin II→AT1 receptor axis (‘bad angiotensin’), inhibits it. As SARS-CoV-2 binds to the ACE2 receptor viral infection therefore disrupts the balance between the two and downregulates NO production.
Low levels of NO production contribute to hypertension [Förstemann and Sessa, 2011] and hypertension is one of the two highested comorbidities for COVID-19 disease progression.
NO is synthesised particularly highly in the paranasal sinuses and nasopharynx [Mantel et al., 2020] of the upper respiratory tract, i.e. the location of SARS-CoV-2’s binding to apical ciliated epithelial cells. This is unsurprising since the airway epithelium is the primary point of contact with all respiratory pathogens.
Nitric Oxide As A Viral Countermeasure.
By January 2021 we had concluded that SARS-CoV-2 was all about viral load in the upper respiratory tract and that NO plays a key role in attacking SARS-CoV-2 virions within minutes of initial infection and by destroying it before its replication cycle was complete. Its SARS-CoV-2-specific functions include inhibition of the RBD:ACE2 through palmitoylation of the receptor binding motif; release from macrophages lysing infected cells; upregulation of mucins to stimulate mucus production (as both a physical barrier in the upper respiratory tract but also to transport virions away from epithelial tissue); degradation of nonstructural proteins nsp3 & nsp4 [Xu et al., 2020] and inhibition of 3-chymotrypsin-like protease (3CLPro, also known as MPro). NO nitrosylates cysteine protease and SARS-CoV-2’s master protease MPro is a cysteine protease. It is necessary to unpack the polyproteins pp1a and pp1ab, to cleave the nsps and – most importantly – to oversee the assembly of the replication and transcription complex (RTC). Interring with MPro inteferes with the mRNA transcription and prevents the viral replication cycle.
As the antivirals (they are not vaccines) are rolled-out, an additional importance of NO has emerged as an anti-adhesion mediator in cellular adhesion, specifically leukocyte:endothelial membrane adhesion.
Artificial Infection Via Injection vs Natural Infection.
Whereas natural infection with SARS-CoV-2 causes the target cellular binding and intracellular fusion in apical, ciliated epithelial cells in the upper respiratory tract, the intramuscular/intradermal delivery of antivirals injects the treatment candidate into the endothelium. This situation – the introduction of the spike protein into the circulatory system – only occurs with natural infection where viral load is so high that viral infection overwhelms the capability of the innate immune response. Such high levels of viral load are principally caused by confining individuals in close proximity to the same other individuals for sustained periods of time (lockdown) and by the sustained reinhalation of any infected sputum and/or mucal ejecta (non-surgical face coverings). In these events, at the end of the week one of the infection cycle, rather than moving toward viral clearance, the infection moves from the upper to lower respiratory tract.
Artificial infection via injection essentially replicates COVID-19 disease progression in the endothelium that would not otherwise occur in 99.6% of individuals, as mRNA experimental ‘vaccines‘ BNT126b2 and mRNA-1273 invite host cells to create a spike protein sequence. Except if the treatment candidate binds to endothelial cells, it will perform this function there. Unsurprisingly, this then triggers an immune response as ‘non-self’ is detected in the circulatory system. The nature of this response, as set out below, presents an increased risk of coagulation and clotting. NO reduces coagulation or clotting risk, hence its crucial importance.
Endothelial Adhesion vs Anti-Adhesion.
The innate immune response to infection includes inflammation and activation of signalling pathways to attract leukocytes to the infection site. In the absence of infection, leukocytes continue in transit but in the event of infection, as they near the infection site their progresion through venules changes. They detect cellular adhesion molecules and begin to slow, moving toward to the endothelial surface. There they begin to be rolled along the surface by binding to the cellular adhesion molecules, particularly selectins P-Selection and E-Selectin as well as ICAM-1 and VCAM-1, in a process unsurprisingly called leukocyte rolling. When at the infection site, the rolling slows to a stop as the leukocytes adhere to the endothelial surface. This is mediated by integrins: VLA-4 ligand on monocytes binds VCAM-1 and MAC-1/LFA-1 ligand (CD11/CD18 complex) on neutrophils binds ICAM-1. At this point, you have leukocyte:endothelial adhesion.
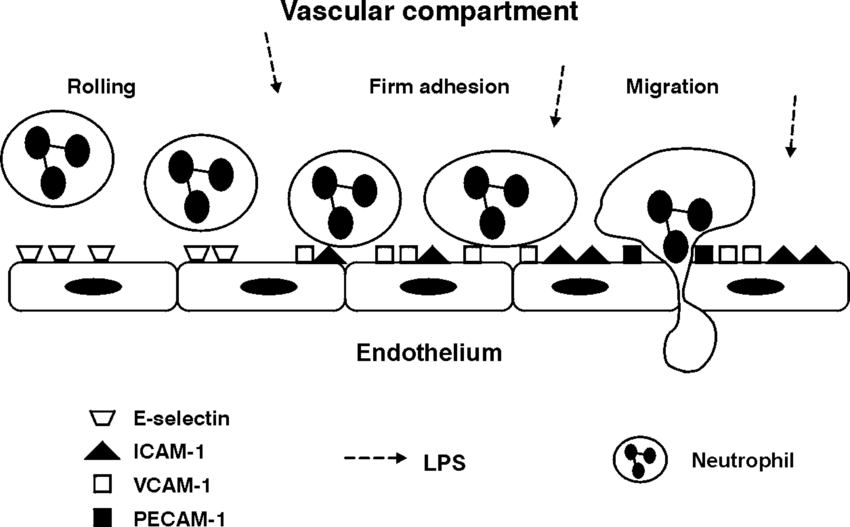
Leukocyte:endothelial adhesion, source: The Airway Compartment: Chambers Of Secrets; Schimmer, Schimmer & Pasch; [2004] Researchgate
The leukocytes are then allowed to pass through the endothelial membrane into the extracellular matrix and on to the actual infection site. This leukocyte recruitment, especially that of neutrophils, is an essential part of the innate immune response. In order to ‘cordon off’ the infected area, platelets are attacted to the site by cytokine and chemokine signalling. Platelets are the cells that cause blood to clot, as a result of clumping together or aggregating. Platelets are recruited to an infection site to seal if off for two reasons: firstly to prevent the spead of the infection – and its causative antigen – away from the infection site and secondly to allow the repair of damaged tissue and any breach in the epithelium.
The three-stage process of rolling, adhesion and endothelial migration produces superoxides, with both the endothelial surface cells and the neutrophils adhered to them using nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) to produce superoxide. Superoxides can also create reactive oxygen species (ROS) that are oxygen-based compounds synthesised as part of the innate immune response, with activated platelets producing ROS. However, at excessive or prolonged levels ROS becomes cytotoxic through oxidative stress.
Platelets show a greater affinity for binding to adhered leukocytes rather than to the endothelial surface itself (to which the leukocytes have been attached), at a ratio of 3:1. This means that they will clump together more within the venule and as they aggregate can form a clot or thrombus. This has the effect not only of constricting and potentially preventing circulation in the venule but also of preventing transit to the localised tissue of other immune cells deployed to fight the infection as well as normal oxygen flow. Platelet:neutrophil complexes further increase ROS production. Activated platelets also shed CD40L – becoming soluble or sCD40L – to act as as signalling molecule to recruit more leukocytes and platelets to the site.
Therefore, both leukocyte:endothelial adhesion and platelet:endothelial interaction can become self-fuelling pro-inflammatory responses. If not controlled, they can cause both acute inflammation (with the resultant risk of excess cytokine signalling) and increased thrombotic risk. Arguably the main control against this is NO.
This is not something new as this has been known for over 20 years, with highlighted studies being Saura et al., [1999]; Keyaerts et al., [2004]; Akerström et al., [2005]; Klingström et al., [2006]; Akerström et al. [2009]; Regev-Shoshani et al., [2013]; Akaberi et al., [2020]; Lisi, Zelikin and Chandrawati [2021].
Conclusions.
- NO is an exceptionally effective and multi-functional viral countermeasure.
- As a vasodilator, NO is an inhibitor of platelet adhesion and subsequent activation. NO is anti-coagulative.
- NO functions as an antagonist to platelet aggregation in response to superoxide as an agonist. In those with vasoconstrictive comorbidities such as hypertension and diet-induced obesity, NO is the anti-inflammatory counterfunction to their chronic pro-inflammatory state. NO is the control response to excessive cytokine release, i.e. cytokine release storm.
- While both NO and superoxide dismutase (SOD) can scavenge superoxide, NO shows a 3-fold increase in non-covalent bonding affinity [Granger and Senchenkova, 2010].
- NO also stimulates soluble SOD1, mitachondrial SOD2 and extracellular SOD3. NO contributes to either maintaining or increasing the shear rate on the endothelial wall and as there is an inverse relationship beetween shear rate and leukocyte adhesion, this reduces initial leukocyte and subsequent platelet adhesion. NO inactivates NADPH oxidase.
- NO is also a defence against the increased risk of thrombophilia triggered by antiphospholipid syndrome mediated by the phospholipid contained in the nanocarriers of BNT126b2 and mRNA-1273,
A healthy level of vitamin D regulates nitric oxide production, with NO serving as an effective viral countermeasure; calcifediol as a potent defence against COVID-19 disease progression & severity and NO as a robust defence against mRNA experimental vaccine-mediated coagulation.