What Variants Want.
Considerable coverage is being given to variant B1.617. This is the ‘Indian’ variant if you use the incorrect basis of giving single country, geographical names to variants that appear globally across multiple variants and are not new, having been in existence for months and in some cases, over a year. As part of their defences against detection, these variants are designed to increase immunoevasion. Like every other virus, SARS-CoV-2 just wants to survive. As a virus it cannot live for long outside of a host cell so its survival is dependent upon it being able to gain continued access to a host cell and then replicate.
Antigen Shift.
The structural spike protein contains the S1/S2 subunits, receptor binding domain and furin cleavage site. This is SARS-CoV-2’s ‘primary weapon’ and is required for cellular binding and then intracellular entry & fusion. To protect this, the structure of the trimeric S protein is subject of continuous shift through its dynamic glycan shielding, with both the position and amount of glycosylation changing.
This shift increases immunoevasion as it provides the S protein with an ‘adaptive camouflage’ or ‘stealth mode’. Antigen shift, the active recombination around the S protein, is one of the main reasons why there has never previously been a successful vaccine or antiviral against coronaviruses. In December 2020, we concluded that “mutation makes a vaccine useless” and our position was validated last month when we evaluated the effect of antigen shift on two of the main antivirals being injected into people – Pfizer/BioNTech’s BNT126b2 and Moderna’s mRNA-1273 – showing that both are highly ineffective against the recurrent amino acid substitutions that are contained within the principal variants.
Our view remains that the S protein is not an effective target for vaccine/antiviral treatments because of antigen shift. It is important to highlight that it can be a target for the adaptive immune’s antigen-specific response because any variants occur before seroconversion. Vaccine/antiviral candidates that target the S protein assume it is static so are defeated by the virus’ defence. The adaptive immune system waits to see what emerges after replication and then reacts accordingly.
Receptor Binding Domain Down.
The receptor binding domain (RBD) that contains SARS-CoV-2’s main epitopes is usually in the ‘down’ position. This means that it is shielded by the S1/S2 subunits and not accessible to the paratopes of circulating antibodies. This can be thought of as a submerged submarine. However, in order to fire it must rise to periscope depth to identify and fire upon its target: a very simple analogy is the raising of the periscope above the surface equates to the raising of one of the three RBDs within the trimeric S protein. In the ‘up’ position, the RBD is primed and can achieve cellular binding, entry & fusion.
This switch from a ‘3-down’ to ‘1-up’ RBD structure allowed amino acid substitution D614G to become the dominant, fixed variant within three months in 2020. This is explained in more detail here.
Positive Sense.
SARS-CoV-2 is a positive sense single-stranded RNA virus. This means that the genetic code contained in its mRNA strand matches human mRNA and so it can be translated directly by the infected host cell without the need for a polymerase.
Interferon Inhibition.
Pattern recognition receptors are contained in cells that form the innate immune system. They are programmed to recognise pathogen-associated molecular patterns of viral RNA, with toll-like receptors TLR7 & 8 and on a good day TLR3 identifying the peptide chains of coronavirus RNA. Two other pattern recognition receptors are retinoic acid-inducible gene I (RIG-1) and melanoma differentiation-associated protein 5 (MDA5).
Upon detection of viral RNA, RIG-1 and MDA5 normally activate and stimulate the interferon signalling pathway. Interferons are a group of cytokine that act both as a first responder and early warning system. Type I interferon (IFN-1) signals to neighbouring cells that an invasive infection is nearby, thereby activating those cells’ own defences. Interferons also act directly on the viral RNA, both to degrade its RNA structure and disrupt its replication [Streicher and Jouvenet, 2019].
Within SARS-CoV-2’s genome, nsp1 & nsp3 contain interferon inhibitors that once unpacked in polyprotein pp1a within the infected host cell, suppress the expression of IFN-1 and therefore the stimulation of interferon-stimulated genes. nsp8 also inhibits activation of MDA5.
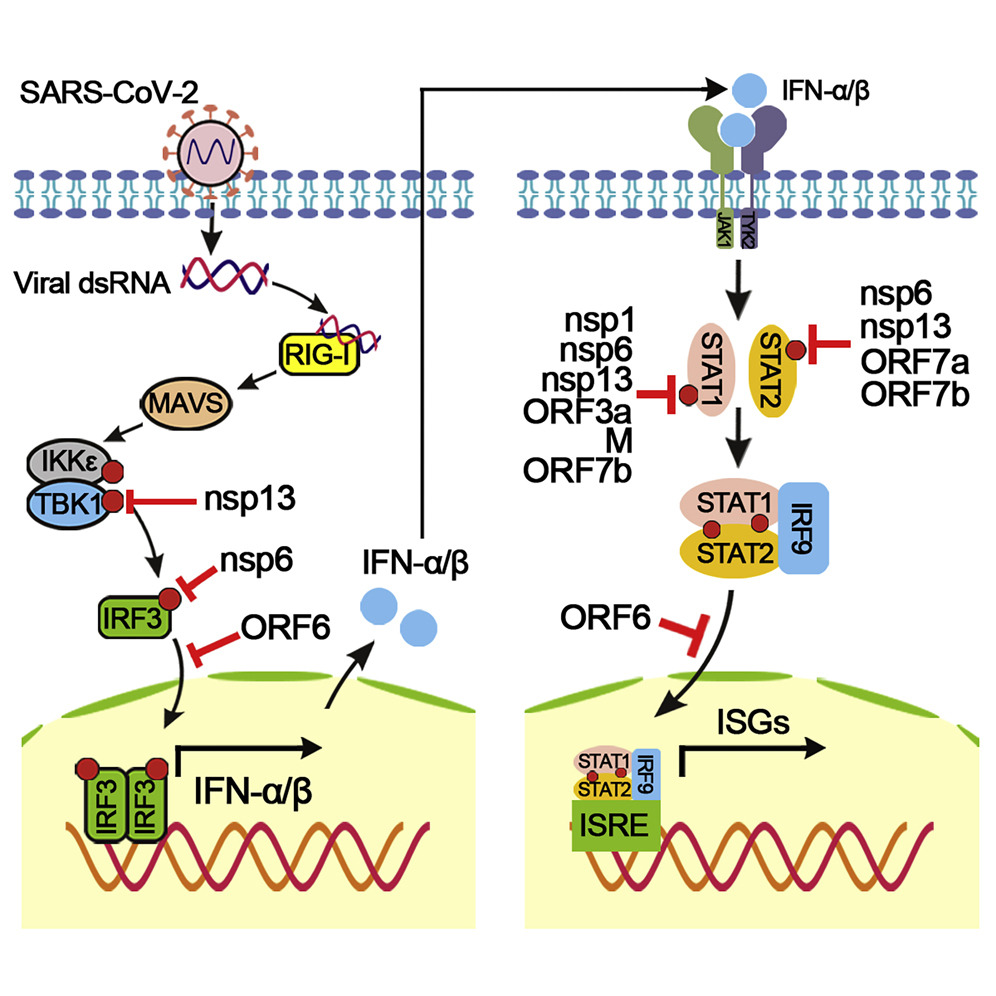
Evasion of Type I Interferon by SARS-CoV-2, Xia et al., 2020. Image courtesy of Sciencedirect.
Myxovirus resistance protein 1 (MxA) is a protein that when expressed targets the infected host cell’s cytoplasm and degrades the virion in the early stage of infection before it begins its unpacking process. Rhinoviruses stimulate MxA but SARS-CoV-2 does not, although a study in March by the University of Glasgow Centre for Virus Research (CVR) showed that co-infection with a rhinovirus and SARS-CoV-2 provided cross-reactivity of MxA. This further supports our view that having a common cold provides an accumulating immunity against SARS-CoV-2 through persistent antigen exposure.
Sa Ribero et al. [2020] show that the combination of the down-regulation of IFN-1 and up-regulation of inflammatory cytokines IL-1β and IL-6 are key factors in any COVID-19 disease progression, particularly in respect of acute respiratory distress syndrome (ARDS).
RNA Capping.
SARS-CoV-2’s RNA uses a 7meGppp (m7G) in the 5′-end of its RNA and 2′-O-methylations at its 5′-terminal nucleotides. The methylation changes the capping from a cap-0 to cap-1 structure [McCaffery, 2019] and mimics human mRNA in the infected cell and therefore bypasses detection by pattern recognition receptors as SARS-CoV-2’s RNA appears as ‘self’ rather than ‘non-self’.
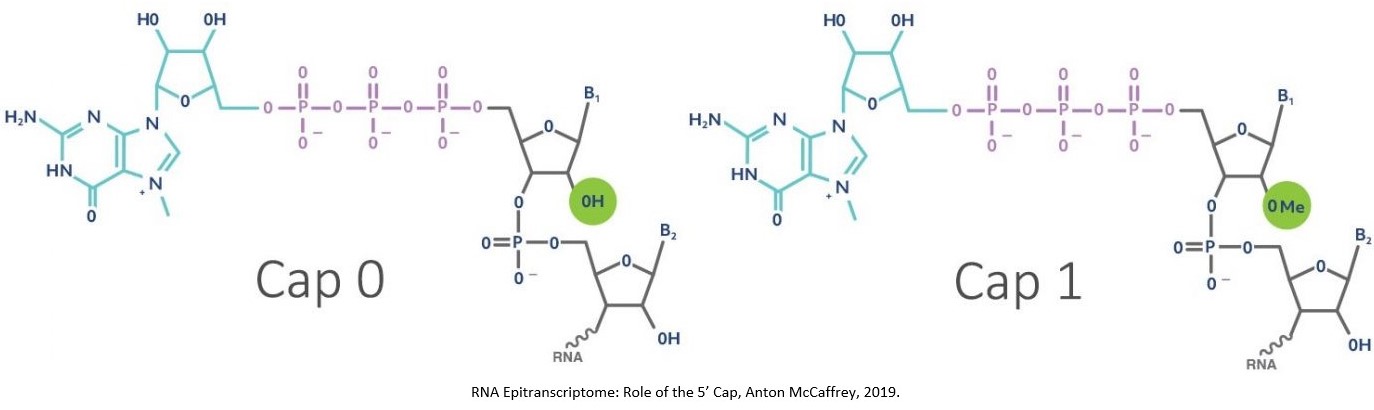
The cap-1 structure is a defence that allows the viral RNA to evade detection by RIG-1 as well as interferon-induced protein with tetratricopeptide repeats (IFIT) proteins. IFIT-1 recognises cap-0 (so RNA without 2′-O-methylation) and binds to its 5-end, blocking the ability to translate the viral RNA and thereby interrupting replication.
S Protein Intercellcular Binding.
Following replication within an infected host cell, the new virions are put together in the endoplasmic reticulum–Golgi intermediate compartment (ERGIC), where the structural S protein is assembled. The virions then move into the Golgi where they are carried by vesicles to the infected host cell are ejected through exocytosis. However, coronaviruses retain a proportion of the newly-replicated S proteins and they are sent directly to the membrane of the infected cell to bind with neighbouring cells. This cell-to-cell fusion creates clusters of cells with multiple organelles, allowing greater viral replication.
This defence is based upon not sending new virions into the humoral system where they will be detected by antibodies and Th2 cells. It is in effect a Plan B, ‘if anything should happen to me, then…’.
Conclusions.
A virus needs a host cell to survive and replicate. The better a virus’ natural defences, the better it can evade detection. The better it can evade detection, the greater its chances of survival. In this respect, evolving effective defences against the immune system is part of natural selection and so it is no surprise that coronaviruses have developed effective defences in the approximately 7,160 years since the beta genus phylogenetically evolved from the alpha genus.
However, the inverse position also applies: the immune system has had the same period of time to react to coronaviruses. As gnathostomes or jawed vertebrates, we first evolved over 500 million years ago and our immune system has been developing over this entire period. While it is facile to state that 500,000,000 is much bigger than 7,160 so humans win every time, this period of evolution and the multi-layered, inter-related nature of our immune system means that while SARS-CoV-2 is clever, it still has weaknesses.
While its cap-1 structure may evade RIG-1, MDA5 and IFIT-1, SARS-CoV-2 antigen is still detected by the toll-like receptors and presented by MHC-I and MHC-II molecules.
SARS-CoV-2 is highly susceptible to vitamin D, with the vitamin D pathways providing both immunity from viral infection and significant attenuation of any disease progression & severity.
The homology of antigen peptide chains across principally other common cold-causing coronaviruses (HCoV-NL63; HCoV-229E; HCoV-HKU1 and HCoV-OC43) but also across other respiratory viruses means that regular exposure to other viruses increases the effectiveness of the innate immune response. Persistent antigen exposure increases immunity as it trains the innate system to respond to other, similar viral infections.
Within the adaptive immune system, memory CD4 TH and CD8 TC cells – both TCM and TEM – conserved from having had a common cold previously, recognise SARS-CoV-2 and upon any primary infection mount an adaptive response as if they have seen it before.
SARS-CoV-2 has several defence mechanisms against detection by the immune system. However the complexity of the immune system means that it can still be detected and the immune system has shown itself to be exceptionally good at overcoming coronavirus infection. There are still risk factors that should be considered, notably high viral load and the impact of comorbidities in disease progression & severity.
To someone with a healthy immune system and without dysfunction in their renin-angiotensin system, SARS-CoV-2 continues to pose a lower risk than seasonal/winter flu (H1N1/H3N2). The far greater risk remains incorrect government strategy and response, as well as the failed NHS clinical response.
The most effective ‘vaccine’ remains your own immune system.